Background
Not long after the successful startup of the MSRE, greater attention was turned to designs for future molten-salt breeder reactors (MSBRs) that would use thorium fuel. Little had been done on future breeder reactor design during the construction of the MSRE, although a 1964 progress report contained a section by Beecher Briggs that described current ORNL thinking on breeder reactors.
The effort to define a molten-salt breeder reactor was driven in large part by the desire to define a follow-on experiment to the MSRE, which ORNL internally called the “Molten-Salt Breeding Experiment,” or MSBE. In order to define the technological goals of an MSBE, it would first be necessary to define an MSBR. Therefore the design team began devoting more and more effort to this reactor concept.
The result of their analysis was described in ORNL-4528, Two-Fluid Molten-Salt Breeder Reactor Design Study. It was a 1000-MWe plant that had four individual reactor units, two of which are depicted in Figure 1. Each provided approximately 550 thermal megawatts of heating power to a series of steam generators. The steam from each reactor module fed a single large 1000-MWe steam turbine.
The motivation for smaller, modular reactor units was a desire to achieve a high capacity factor. Individual units could be taken offline and repaired or maintained while the plant itself continued to operate at a fractional electrical output.
Two modular reactor units with their heat exchangers and steam generators, described in ORNL-4528. The flow channels for the fuel salt are colored in dark blue, the blanket salt in green, hot coolant salt in light purple, and cooler coolant salt in dark purple. The steam channels are shown in light blue.
Cutaway view of the interior of one of the modular reactor units described in ORNL-4528. The reactor has three regions: the core region, shown in blue, consists of hexagonal graphite prisms with internal recursive channels for the flow of fuel salt. The blanket region, shown in green, consists of simple graphite cylindrical channels open at each end, filled with blanket salt. The reflector region, shown in dark grey, consists of cylindrical extrusions of graphite. The entire structure is contained in a vessel made of Hastelloy-N. The core region is attached to a plenum structure at the base of the reactor which collects salt as it enters the reactor and as it leaves.
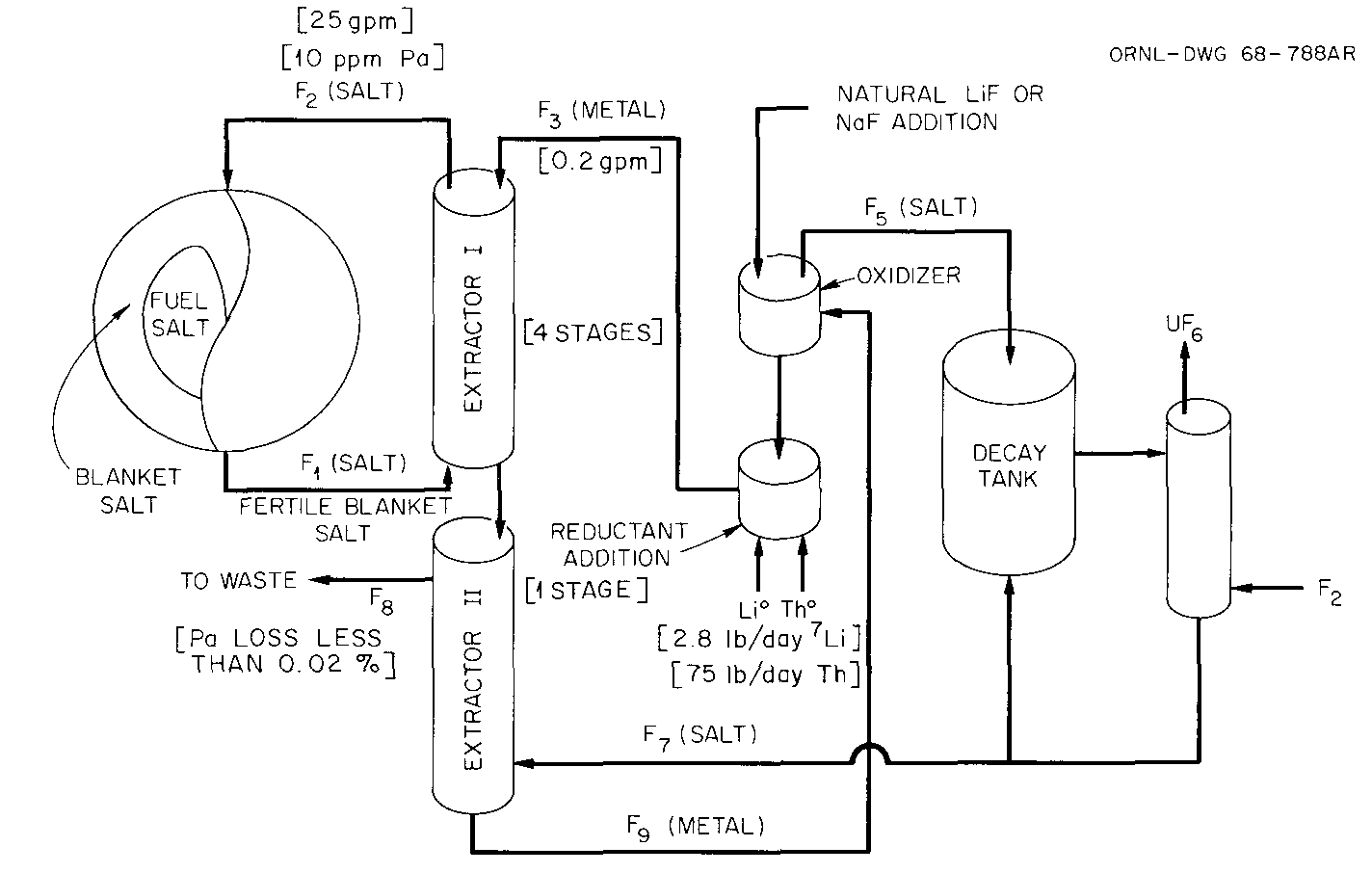
Chemical Processing
The chemical processing system for the reactor is shown in Figure 3. In the reactor, nuclear fission reactions take place that heat the salt and release high-energy neutrons. These neutrons are slowed down by collisions with the atoms of the graphite moderator to increase the probability that they will cause other reactions. About half of these neutrons are absorbed by the thorium in the blanket salt of the reactor, which consists of highly-depleted lithium fluoride (HDLiF), beryllium fluoride (BeF2), and thorium tetrafluoride (ThF4).

As thorium absorbs neutrons, it rapidly transmutes into 233Pa, which has a 27-day half-life, decaying into 233U. It is very undesirable that 233Pa absorb another neutron before it decays to 233U, since that would cause it to transmute into 234U, which is not a reactor fuel. Therefore, ORNL engineers and chemists felt that it would be important to remove protactinium from the blanket of the reactor along with any uranium that had already formed from the decay of protactinium. They anticipated using a technique called “reductive extraction” to accomplish this removal. It involved introducing a stream of the blanket salt (containing very small amounts of protactinium and uranium) into the bottom of a extraction column. At the top of the same extraction column, labeled “Blanket Extractor” in Figure~??, a stream of liquid metallic bismuth would have been introduced containing 3000-4000 ppm of metallic thorium. Contact between the metallic thorium and the protactinium and uranium tetrafluorides dissolved in the salt led to an exchange of fluoride ions. First uranium and then protactinium would have been reduced from tetrafluorides to metals while the thorium in the bismuth was oxidized to a tetrafluoride. The oxidized thorium entered the blanket salt while the reduced uranium and protactinium entered the bismuth stream. Thorium was an ideal reductant because the removed protactinium was replaced by an equivalent amount of the fertile material. ORNL researchers estimated that 96% of the protactinium and uranium present in the blanket would be removed by this technique, and that which was not removed was not lost; it simply stayed in the blanket fluid and was removed on another chemical processing pass.
The bismuth stream, containing small amounts of dissolved metallic protactinium, uranium, and unused thorium reductant, was directed towards a hydrofluorination column, where the metallic Th, Pa, and U were oxidized to tetrafluorides in the presence of a “decay salt” mixture, likely a very similar combination to that used in the blanket salt. The decay salt was routed back to the decay tank, where 233Pa is given time to decay to 233U outside of the neutron flux of the reactor. The bismuth stream emerging from the hydrofluorinator was loaded with new metallic lithium and thorium to serve as reductants.
To remove decayed uranium from the decay salt, fluorination with molecular fluorine (F2) was proposed. Fluorination would promote UF4 to UF6, but would leave protactinium behind. Thus it represented a simple technique to separate the desirable uranium product from protactinium which still needed time to decay. The UF6 generated in the column marked “Decay Fluorinator” in Figure~?? was then directed to a reduction column, where the UF6 was directly reduced to UF4 in the presence of LiF-BeF2 salt that had been purified in a distillation column.
Fuel salt processing involved fewer chemical processing steps. Fuel salt, consisting of lithium fluoride, beryllium fluoride, and uranium tetrafluoride, and containing a certain concentration of fission product fluorides, was first directed to a fluorination column, marked “Fuel Fluorinator” in Figure~??. The fluorination process had to be very efficient to prevent excessive losses of uranium to the next process. UF6 generated through fluorination was directed to the same reduction column as UF6 that had been generated from the fluorination of decay salt.
The fuel salt, essentially stripped of uranium, passed into a distillation vessel where it was heated to temperatures of 1000°C at very low pressures. In these conditions, LiF and BeF2 boiled out of the salt and were condensed on the top interior surface of the still, then directed to a collection region and pumped to the reduction column. Fission product fluorides, which did not boil off in this process, collected at the base of the distillation unit and were periodically tapped off for transfer to another area where they were prepared for disposal.
The purified LiF-BeF2 and the UF6 generated through fluorination were recombined into a fuel salt by passing hydrogen gas through the reduction column. Hydrogen reduced UF6 to UF4, generating HF in the process, which was then used in the hydrofluorinator as the reactant.
Using these processing techniques, it was anticipated that thorium could be introduced to the system, first as a reductant in bismuth. Since the amount of reductant far exceeded the minimum amount needed to reduce protactinium and uranium, a great deal more thorium would be used than was actually consumed in the reactor’s blanket. An alternative process would have utilized an electrolytic cell to electrolyze ThF4 in the presence of bismuth, creating metallic thorium dissolved in bismuth and bismuth trifluoride (BiF3). But BiF3 was a very corrosive agent to the structural materials that might be employed in the system. This alternative approach was depicted in Figure~??.
With thorium metal supplied by an electrolytic cell rather that directly supplied, an individual thorium atom would likely undergo multiple passes through the extraction column (oxidized to salt in the hydrofluorinator and reduced back to metal in the electrolytic cell) before it first entered the blanket salt by being oxidized to salt while it reduced a protactinium or uranium ion to metal. In the blanket, thorium would eventually absorb a neutron, forming protactinium which would be removed in the extraction column by reduction to metal in bismuth. That metallic protactinium would be oxidized to protactinium tetrafluoride in the hydrofluorinator and pass into the decay tank. After undergoing radioactive decay to 233U, the newly-formed uranium tetrafluoride would be removed from the decay tank by fluorination to UF6. The UF6 would pass to the reduction column, where it would be reduced to the tetrafluoride through contact with hydrogen, but this time it would be a part of the fuel salt and pass back into the reactor.
Exposed to neutrons, ultimately the 233U would fission into two fission products, each of which would rapidly form fluorides from the four fluoride ions released in the fission of uranium tetrafluoride. Depending on the chemical nature of the two fission products, one or the other might pass out of the fuel salt as a noble gas or be collected on an interior surface as a noble metal. But if the fission product was an alkali metal, like rubidium or cesium, or an alkaline earth metal, like strontium or barium, or a lanthanide like cerium, lanthanum, neodymium, or samarium, it would be removed from the reactor when a stream of fuel passed into the distillation column and the LiF-BeF2 solvent was boiled away from the fission products.
Hence, a thorium atom enters the bismuth extraction column and ultimately two fission products are removed from the reactor as a noble gas, a noble metal, or as the distillate at the base of the distillation column.

Fluorination Experimental Development
Fluorination converts the uranium tetrafluoride in the salt to volatile hexafluoride according to the reaction:

Since some fission and corrosion products (Mo, Ru, Nb, Cr, Te, and Tc) also form volatile fluorides, the gases were to be passed through traps of MgF2 and NaF pellets at 400°C. These sorbers would retain the contaminants while passing UF6, which would have been subsequently collected in a cold trap at -70°C for recycle to the reactor fuel stream.
Previous experience with batch fluorination of UF4 in molten salt showed that nearly complete volatilization of UF6 could be obtained at reasonable rates by sparging the salt with fluorine. The major problem associated with making this step continuous was corrosion, for which a frozen layer of salt on the vessel wall appeared to be the best strategy for mitigation.
Tests on a batch frozen-wall fluorinator were conducted in support of the molten-salt fluoride volatility process. Internal heat generation was provided by resistance heating, using nickel electrodes and 60-cycle AC power. This system was operable with gas flowing through an unheated line that entered the fluorinator at a point below the surface of the molten salt. Electrodes would not be necessary for a fluorinator operating on the fuel stream from an MSBR since adequate internal heat generation will be provided by decay of fission products in the salt.
For the reference 1000-MWe reactor it was necessary to cool the fuel stream for 1.5 days to reduce the specific heat generation rate to 3 x 104 BTU/(hr-ft3) to facilitate temperature control.
Further studies of continuous fluorination were undertaken in 1966 using a 1-in.-diam nickel column having a salt depth of 48 in. No provision was made for corrosion protection by a frozen layer of salt and corrosion rates were, as expected, quite severe. Fluorination tests in which molten salt (NaF-LiF-ZrF4) containing 0.5 wt % UF6 was contacted countercurrently with fluorine at 600°C demonstrated removal of uranium from the salt with 96 to 99.4% efficiency in a 1-hr period of continuous operation. The flow rates (cc/min) for the molten salt and the fluorine were 15 and 70 (STP) respectively. Material balances were complicated by the inevitable corrosion of the nickel vessel. Complete removal of uranium from the salt with no corrosion would yield, for the above conditions, a UF6 concentration of 17.6 mole% in the off-gas. Observed concentrations ranged as high as 35 mole% UF6.
In 1967, experimental studies of continuous fluorination of molten salt were made in a system consisting of a 1-in.-diam, 72-in.-long nickel fluorinator and auxiliary equipment (Figure~??), which allowed the countercurrent contact of molten salt with fluorine. The fluorinator off-gas passed through a 400°C NaF bed for removal of chromium fluorides, a 100°C NaF bed for removal of UF6, and a soda lime bed for F2 disposal. A gas chromatograph was used to analyze the off-gas for F2, UF6, and N2 just prior to its passage through the 100°C NaF bed. The uranium concentration in the salt after fluorination was determined from salt samples.
\begin{figure}[htp]
\begin{center}
\includegraphics[width=\textwidth]{images/ORNL4145_fig3-1.png}
\end{center}
\caption{Equipment for Removal of Uranium from Molten Salt by Continuous Fluorination.}
\label{4145-fig:3-1}
\end{figure}
Fluorination tests were made in which molten salt (41.2-23.7-35.1 mole% NaF-LiF-ZrF4) containing UF4 was contacted countercurrently with a quantity of fluorine in excess of that required for conversion of UF4 to UF6. During a given experiment, salt and fluorine feed rates, operating temperature, and UF4 concentration in the feed salt were maintained constant. However, these parameters were varied from one experiment to the next, as follows: operating temperature, from 525 to 600°C; salt feed rate, from 5 to 30 cm3/min; fluorine feed rate, from 75 to 410 cm3/min; and UF4 concentration in the feed salt, from 0.12 to 0.35 mole% UF4.
\begin{figure}[htp]
\begin{center}
\includegraphics[width=\textwidth]{images/ORNL4145_fig3-2.png}
\end{center}
\caption{Variation of Uranium Removal with Salt Throughput, Operating Temperature, and UF4 Concentration in Feed Salt.}
\label{4145-fig:3-2}
\end{figure}
The effects of salt throughput, operating temperature, and initial UF4 concentration on uranium removal during steady-state operation are shown in Figure~??. The data were based on the average uranium concentration in the fluorinated salt, which was determined at 15-min intervals during 1- to 2-hr periods of steady-state operation. A salt depth of 48 in. was used in the fluorinator in all tests, and the fluorine feed rate was varied from 215 to 410 cm3/min (STP). Removal of the uranium fed to the fluorinator ranged from 97.4 to 99.9%, with removal in most of the runs being greater than 99%. Uranium removal was observed to decrease as the salt throughput was increased, as the operating temperature was lowered, and as the UF4 concentration in the feed salt was decreased. As long as the quantity of fluorine used was stoichiometrically adequate, no significant effect of fluorine feed rate was noted.
In 1968, the feasibility of forming and maintaining a layer of frozen salt (which will protect the walls of a continuous fluorinator) was established by laboratory experiments using a countercurrent flow of molten salt and an inert gas. The experimental equipment consisted of a 5-in.-diam by 8-ft-high column, fabricated from sched 40 nickel pipe (Figure~??). An internal heat source consisting of three Calrod heaters contained in a 3/4-in.-diam sched 40 Inconel pipe was used to simulate the volume heat source that would be provided by fission product decay in the molten salt. Two sets of internal thermocouples, located near the center of each of two test sections, measured the radial temperature gradient. The location of the interface between the molten and the frozen salt could then be established from these measurements. Each test section was independently cooled by air flowing through spirally wound 3/8-in.-diam nickel tubing. Additional Calrod heaters were wound on the external surface of the fluorinator to provide auxiliary heat during heatup and to provide temperature control at the ends of the column. A 66-34 mole% LiF-ZrF4 mixture, which has a liquidus temperature of 595°C and a phase diagram similar to the LiF-BeF2 system, was metered from the feed tank for periods as long as 5 hr. This allowed data to be collected for a 1- to 2-hr period of steady-state operation.
\begin{figure}[htp]
\begin{center}
\includegraphics[width=0.5\textwidth]{images/ORNL4272_fig1-1.png}
\end{center}
\caption{Experimental Equipment for Studying the Formation of a Frozen Salt Layer for Corrosion Protection. All external surfaces were provided with electrical heaters and were heavily insulated.}
\label{4272-fig:1-1}
\end{figure}
The principal objective of the experiments (i.e., the demonstration that a layer of frozen salt can be formed and maintained under approximate operating conditions) was achieved. In Table~??, experimental conditions are compared with reference conditions for processing the fuel stream of a 1000-MWe MSBR; the fluorinator has a salt throughput of 15 ft3/day and an inlet uranium concentration of 0.8 kg per cubic foot of salt, with a 50% fluorine utilization.
\input{tables/ORNL4272-table-1-1}
In general, the thicknesses of the frozen wall and the temperature profiles in the frozen salt were in good agreement with the values that were obtained by assuming radial heat transfer from a volume heat source. The thickness of the frozen wall ranged from 0.3 to 0.8 in., depending on operating conditions. The effect of heat generation in the layer of frozen salt was not simulated in these experiments.
The thermal conductivity of the frozen salt was calculated for each run from the experimentally determined temperature gradient and the measured heat flux; the relative agreement of calculated values was assumed to be indicative of the consistency of the experimental data. Thermal conductivity values calculated from the upper test section data were closely grouped around 0.75 BTU/(hr-ft-°F); however, values from the lower section were more widely scattered and were generally about 100% higher.
The heating-cooling system used on the column produced some variation in the temperature of the external wall (and hence the thickness of the frozen wall); in a typical run, the difference between the temperature of the salt liquidus and that of the wall ranged from 85 to 140°C.
Protection of the fluorine inlet nozzle from corrosion was an anticipated problem that was associated with operation of a frozen-wall fluorinator. A possible solution to this problem consisted of introducing the fluorine through a short section of 3-in.-diam pipe that intersects the fluorinator at a 45° angle. The inlet section would be protected from corrosion by a layer of frozen salt as in the fluorinator. Results of tests indicated satisfactory operation when the surfaces of the inlet section were covered by a layer of frozen salt that was produced by maintaining wall temperatures below the temperature of the salt liquidus. Heat was being supplied to the inlet section by means of turbulence (caused by bubbles) in the molten salt; in an actual instance, heat would be generated in the salt as a result of fission product decay. This method of introducing gas appeared to be feasible, although it will not produce small-diameter gas bubbles.
Distillation Experimental Development
In order to support the proposed distillation of the LiF-BeF2 carrier salt from fission products, it was necessary to determine the relative volatilities of the lanthanide fission products relative to lithium fluoride. Ideally, the lower the relative volatility of the fission products relative to LiF, the more effective would be their separation through distillation. This work was reported on in ORNL-3830 in May 1965.
The experimental apparatus used to determine relative volatility included a cold finger to condense the vapor phase in a small vessel containing salt brought to the desired temperature and pressure, as shown in Figure~??.
\begin{figure}[htp]
\begin{center}
\includegraphics[height=0.6\textheight]{images/ORNL3830_fig18-2.png}
\end{center}
\caption{Equilibrium Still with Cold Finger.}
\label{3830-fig:18-2}
\end{figure}
The significant fission product lanthanides were analyzed with the exception of promethium. The results of the analysis are given in Table~??. They show that the average relative volatilities of the trivalent rare earths in LiF varied from 0.01 to 0.05. There appears to be a trend toward decreasing relative volatility with increasing temperature. Cerium tetrafluoride showed a relative volatility of about 0.15. Scouting tests with CsF showed it to have a relative volatility somewhere between 6 and 16.

The following year another experimental apparatus was used to measure relative volatilities, and this work was reported on in ORNL-3945 in May 1966.
The volatility data was measured with a recirculating vapor-liquid equilibrium still shown in Figure~??. It was not as simple to operate as the cold-finger apparatus, but it was not subject to the same experimental biases. Vapor was continuously generated in the 2-in. still pot, condensed in the 1-in. condenser, and returned as liquid to the still pot. When concentrations reached a steady state, the concentration of the liquid in the still pot and that of the liquid in the condenser were equilibrium values from which relative volatilities could be determined, provided the liquid volumes in the condenser and boiler sections are well mixed. Measurements were made by freezing the salt and cutting the still in pieces to take samples.
\begin{figure}[htp]
\begin{center}
\includegraphics[height=0.7\textheight]{images/ORNL3945_fig3-2.png}
\end{center}
\caption{Schematic Diagram of Molten-Salt Equilibrium Still.}
\label{3945-fig:3-2}
\end{figure}
Several runs lasting from 2 to 30 hr were made with this still at 1000°C and 0.5 mm Hg. Relative volatilities for systems of LiF containing from 1 to 2% selected rare-earth fluorides were measured. The relative volatility of cerium apparently lay between 0.0014 and 0.003; the relative volatility of neodymium lay between 0.00055 and 0.00089; and lanthanum had a relative volatility of about 0.00067. These results were reproducible after initial difficulties with still operation were resolved. The principal problems appeared to be: (1) avoiding excessive contamination of the condensate collector when charging the salt to the system, and (2) salt clinging to the walls where it could not be mixed with the bulk of the salt.
The relative volatility of CeF3 determined by the equilibrium still was about one-tenth of that reported previously from cold-finger measurements, and the relative volatilities of NdF3 and LaF3 were one-fiftieth of those previously reported. These more accurate values were low enough to permit comfortable operation of a batch distillation system in the fuel-salt flowsheet.